Developing solutions for missing variant-haplotype information (missing VHI)
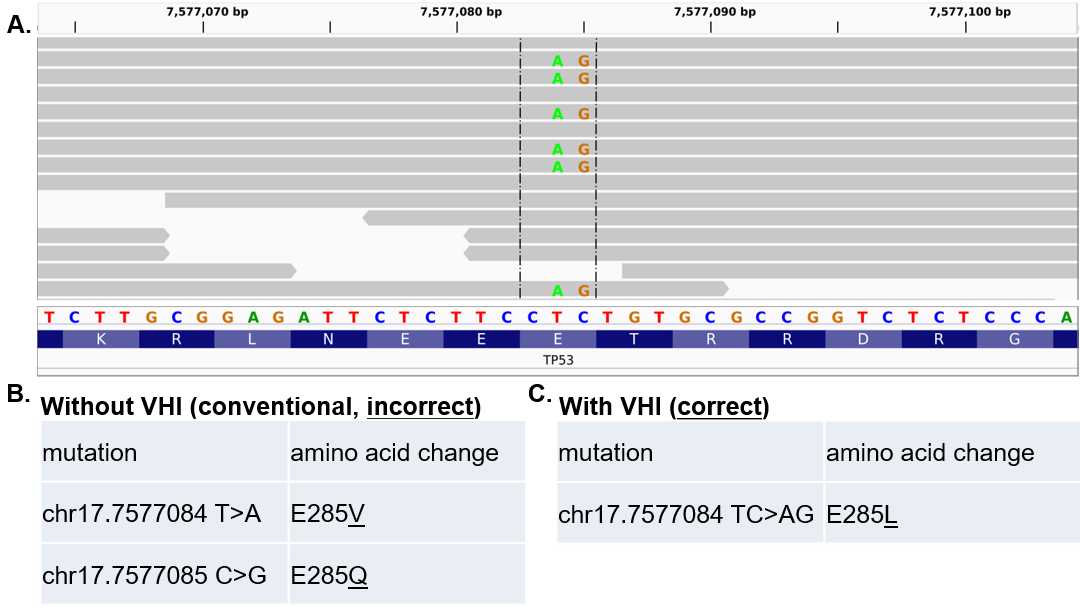
Missing VHI is a fundamental bioinformatics problem caused by the fact that humans have a diploid genome, but most bioinformatics pipelines were developed based on a human reference containing only one set of chromosomes and without VHI retained.
Issues resulting from missing VHI, especially ignoring proximal variants on the same chromosome, can cause many problems for variant annotation and interpretation:
Identifying and fixing incorrect amino-acid-change predictions caused by missing VHI
We developed Multi-Nucleotide Variant Annotation Corrector (MAC):
MAC is the first software for solving a type of missing VHI problem. Since it was published, MAC has been cited in many high-impact papers:
- Wang Q, et. al. Nat Commun. 2020 May 27;11(1):2539
- Łuksza M, et. al. Nature. 2017 Nov 23;551(7681):517-520
- Riaz N, et. al. Cell. 2017 Nov 2;171(4):934-949.e16
Collaborations
- We’re collaborating with Roswell Park’s Dr. Martin Morgan to develop bioinformatics algorithms for solving many types of systematic errors introduced by missing VHI in computational predictions.
- We’re collaborating with Dr. Ailong Ke at Cornell University to develop on-demand computational tools for CRISPR-Cas based genome editing, including identifying therapeutic target and quantifying the risk of off-target effect based on the sequence differences.
Defining negative status for multi-sample comparison using next-generation sequencing
Cancer-related somatic mutations can be measured using next-generation sequencing (NGS). For a specific mutation in a given sample, an NGS test may yield three possible statuses: positive, negative, or unknown mostly due to low coverage.
A common solution is to require the coverage at the site of a mutation to pass a universal minimum coverage (UMC). However, this method relies on an arbitrarily chosen threshold, and does not take into account the mutations' relative abundances.
We propose an adaptive mutation-specific negative (MSN) method to improve the classification of negative and unknown mutation statuses in the comparison of multiple "related" tumor samples of the same patient.
We evaluated the performance of MSN using a real dual-platform single-cell sequencing dataset, the MSN method not only assigned negative statuses with better accuracy than the conventional UMC method, but more importantly rescued more data which would otherwise being classified as “unknown” using UMC.
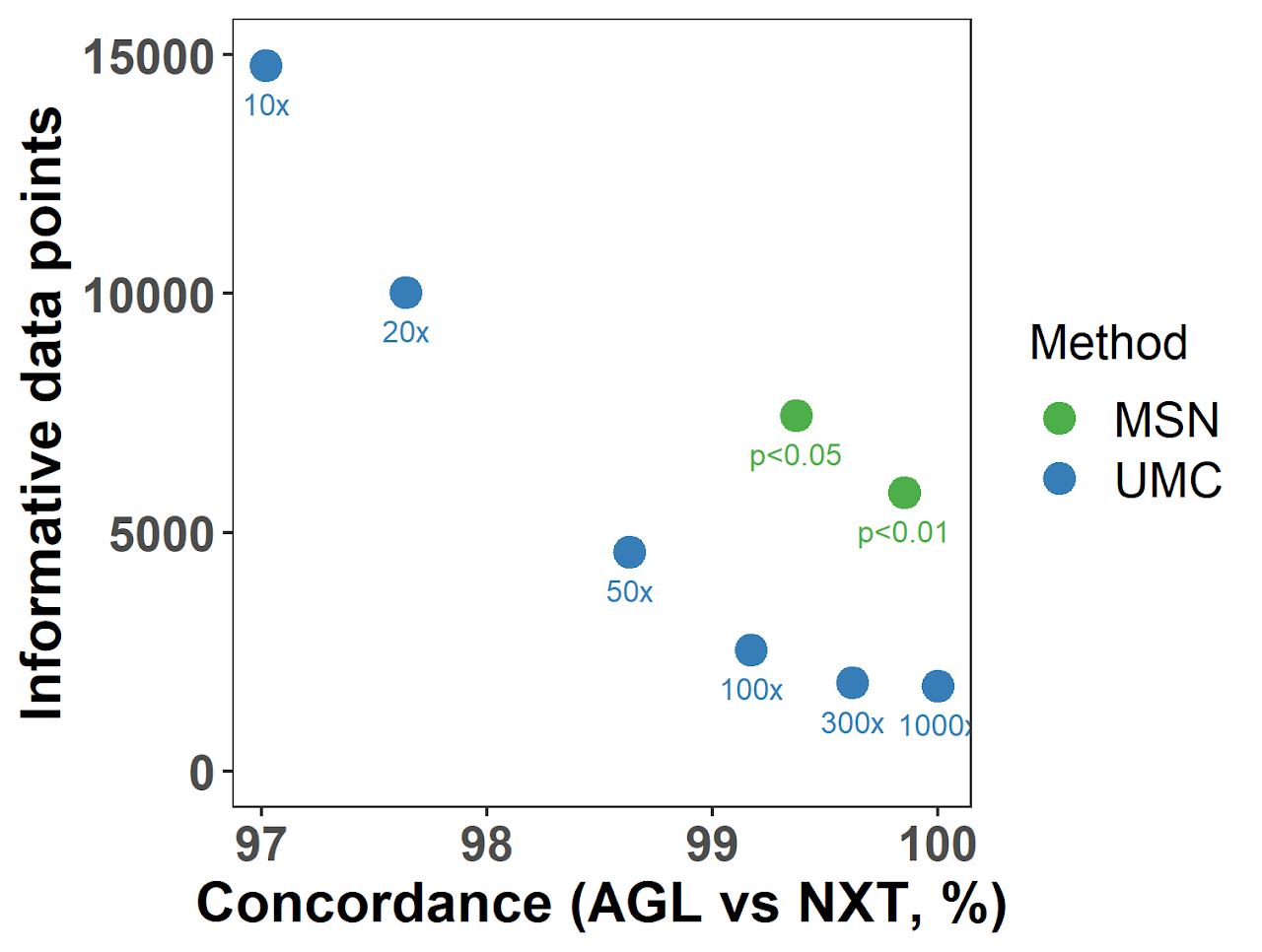
Two negative-defining methods, UMC and MSN, were tested in a single-cell dual-platform sequencing data set using varying thresholds (indicated under each dot). The performance of each run was evaluated by:
- The concordance of mutation statuses between two paired runs (Nextera, NXT and Agilent, AGL) of the same single cell (X-axis); and
- The total number of informative data points after excluding unknown statuses (Y-axis).
To the best of our knowledge, MSN is the first method dedicated to defining negative mutational status. In addition to tumor heterogeneity analyses, this method also works on other samples containing tumor cells or tumor DNA, such as circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA).